Identifying Genes Involved in Alkaloid Biosynthesis in Vinca minor through Transcriptomics and Gene Co-Expression Analysis

 , , , , , , , , , ,
, , , , , , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plant Growth Conditions and Sample Collection
2.3. RNA-Sequencing
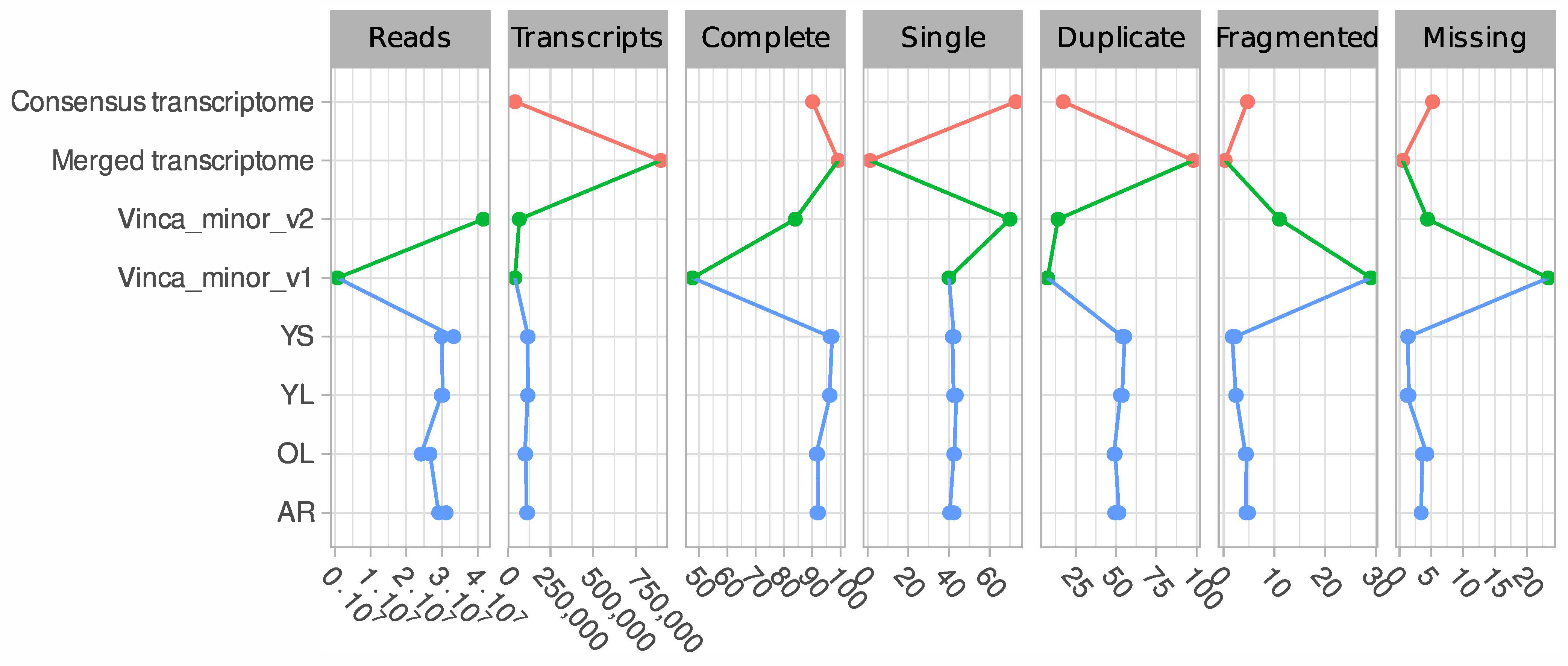
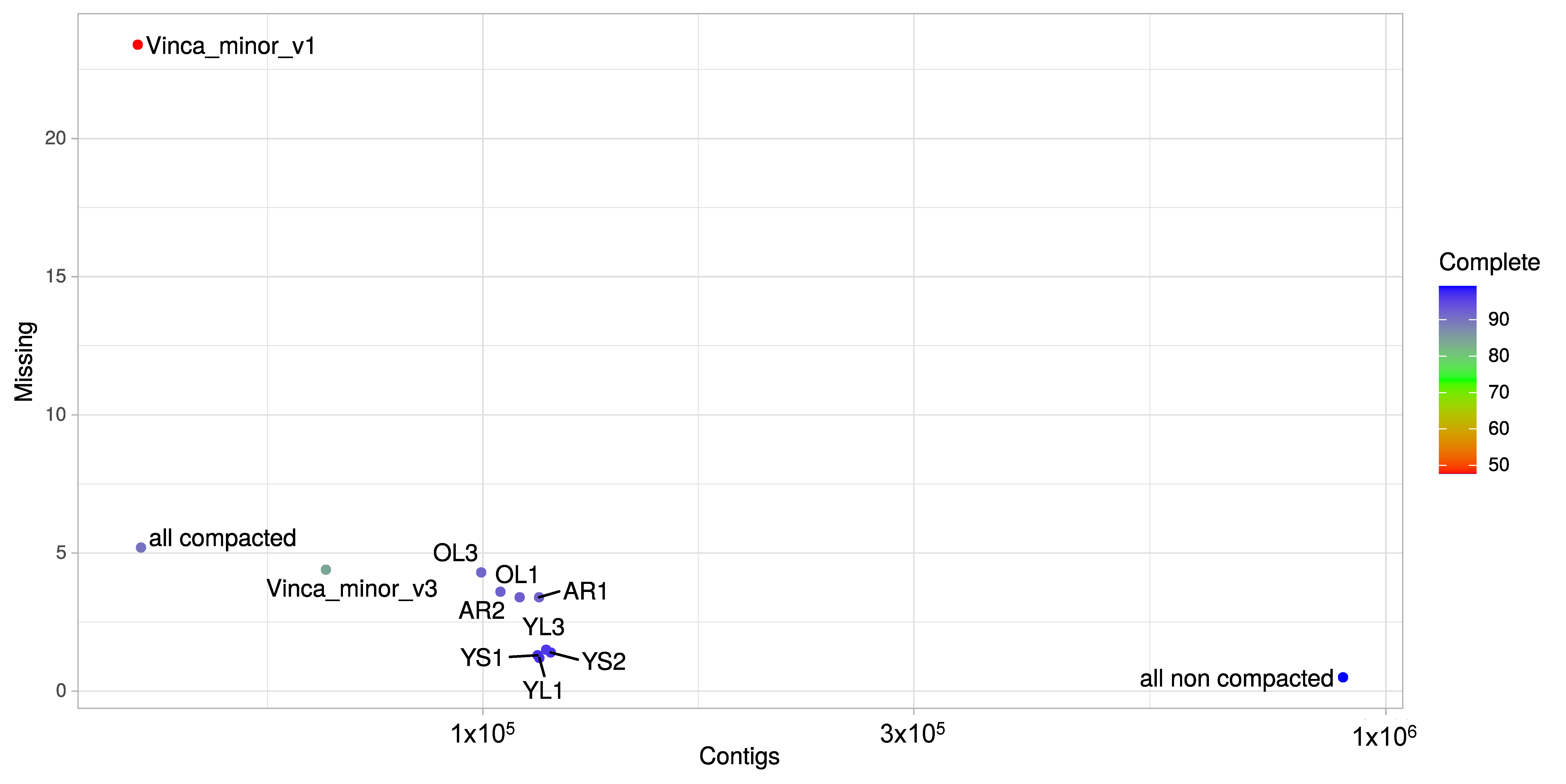
2.4. Generation of a Consensus Transcriptome
2.5. Functional Analysis of the Consensus Transcriptome
2.6. Heterologous Expression of Tabersonine 16-O-Methyltransferase Candidates in Yeast
2.7. Recombinant Vm16OMT Production in E. coli, Substrate Specificity Assays and Kinetic Parameters Determination
2.8. UPLC-MS Analyses
2.9. Gene Expression Measurements Using Real-Time RT-PCR
2.10. Subcellular Localization Studies
2.11. Co-Expression Network
3. Results and Discussion
3.1. A Consensus and Refined Transcriptome Resource for V. minor
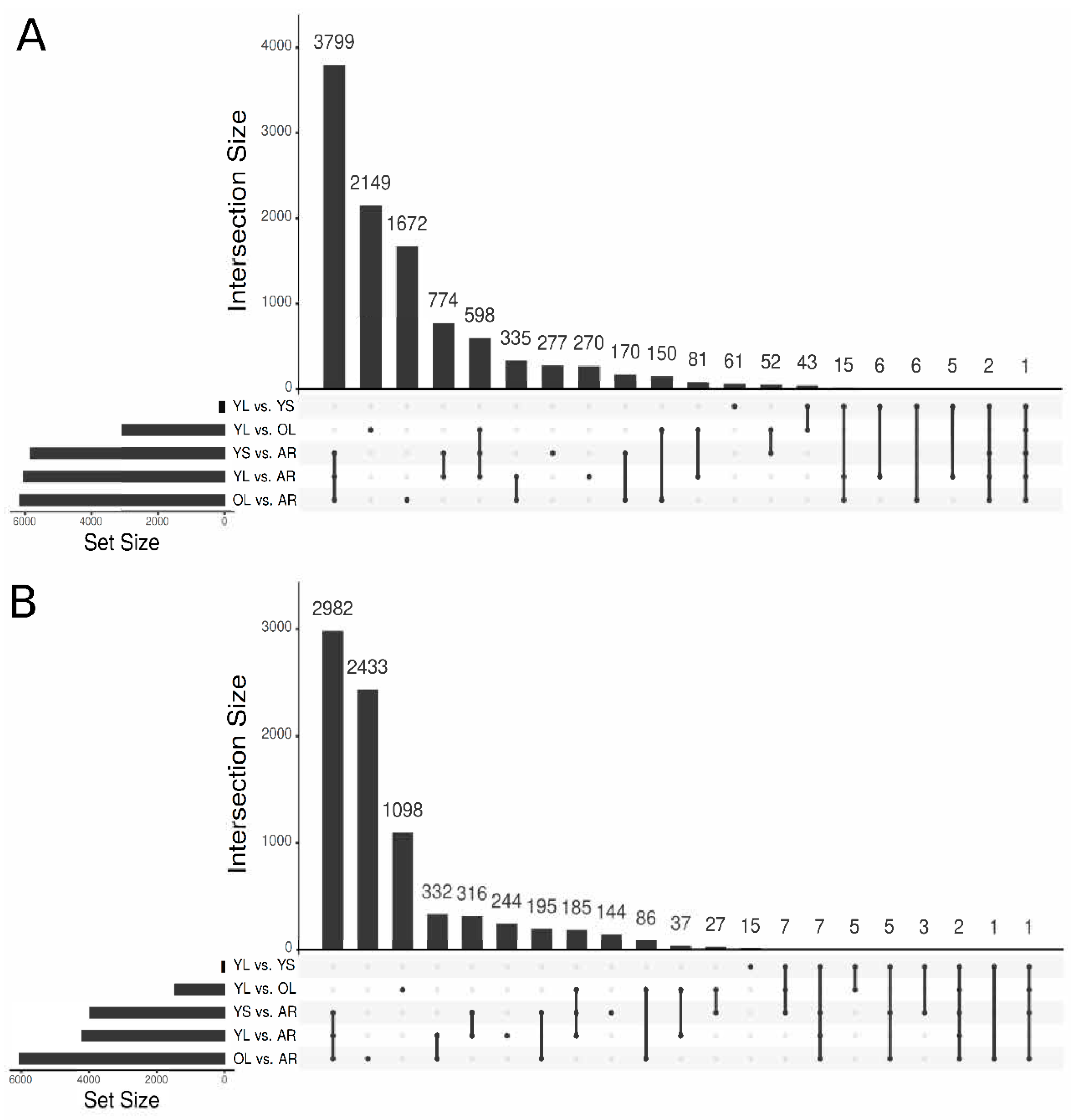
3.2. Functional Annotation of the V. minor Consensus Transcriptome and Differentially Expressed Gene Analysis
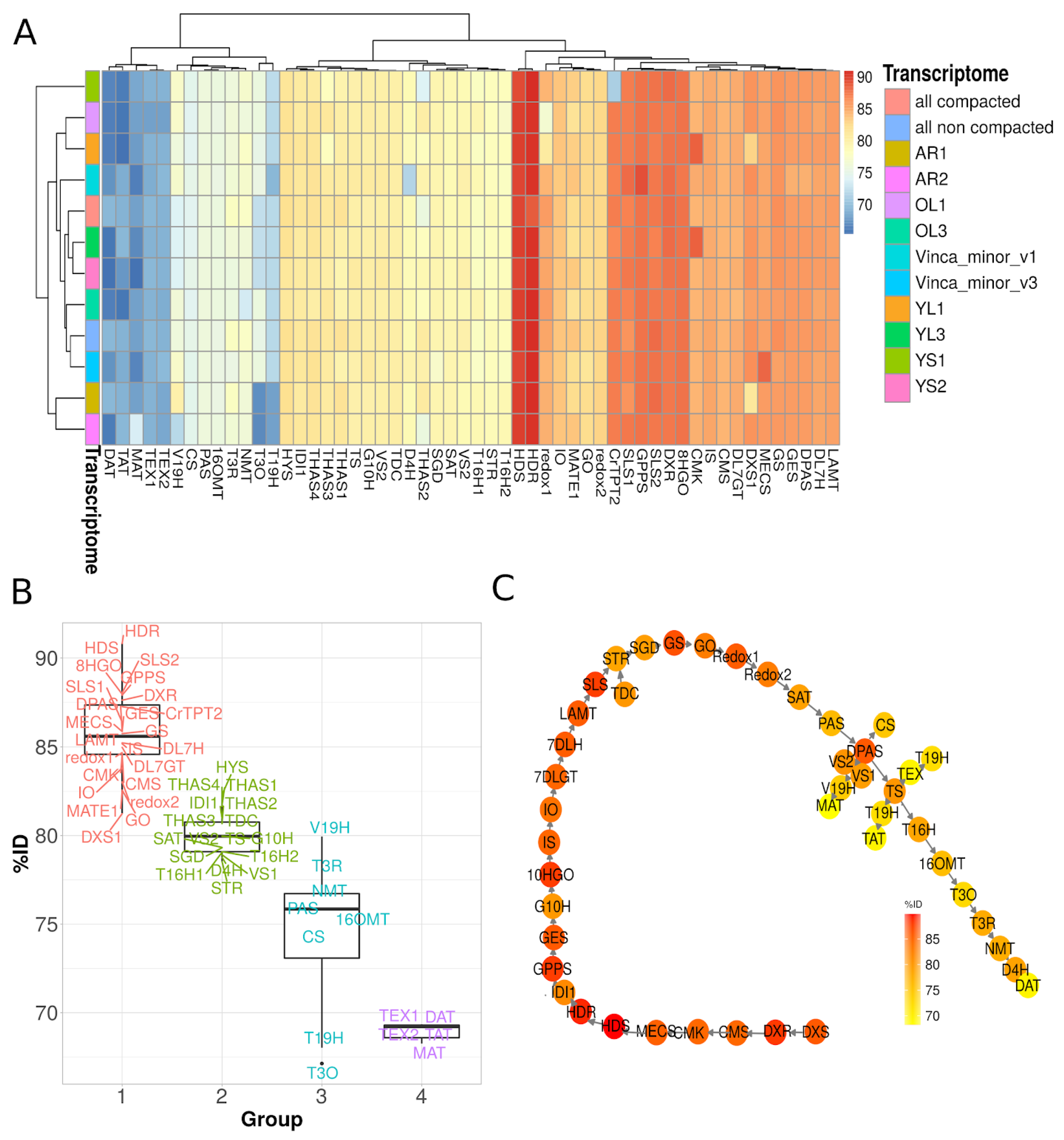
3.3. Identification of Candidate Genes from the MIA Pathway through Homology-Based Predictions
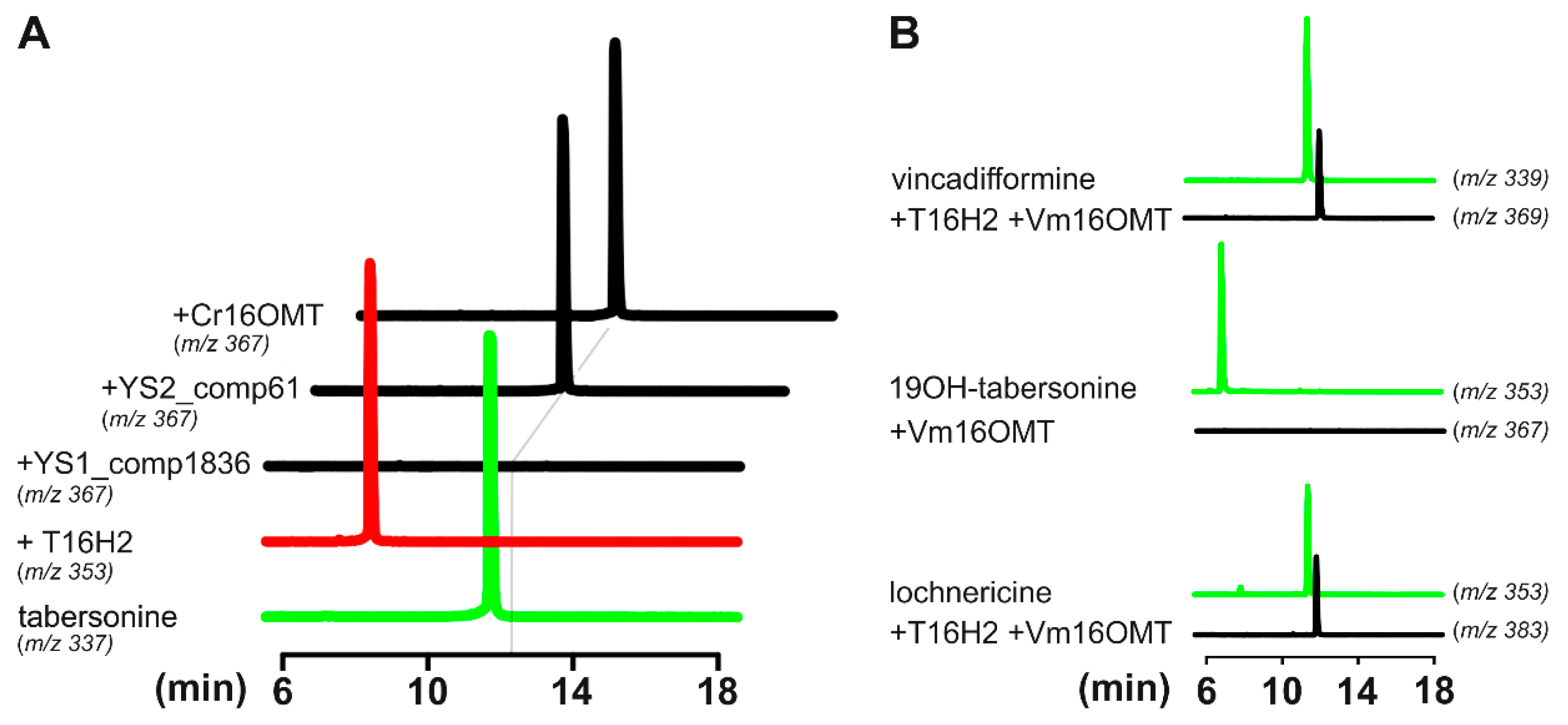
3.4. Identification and Functional Validation of a V. minor Vincadifformine/Tabersonine 16-O-Methyltransferase
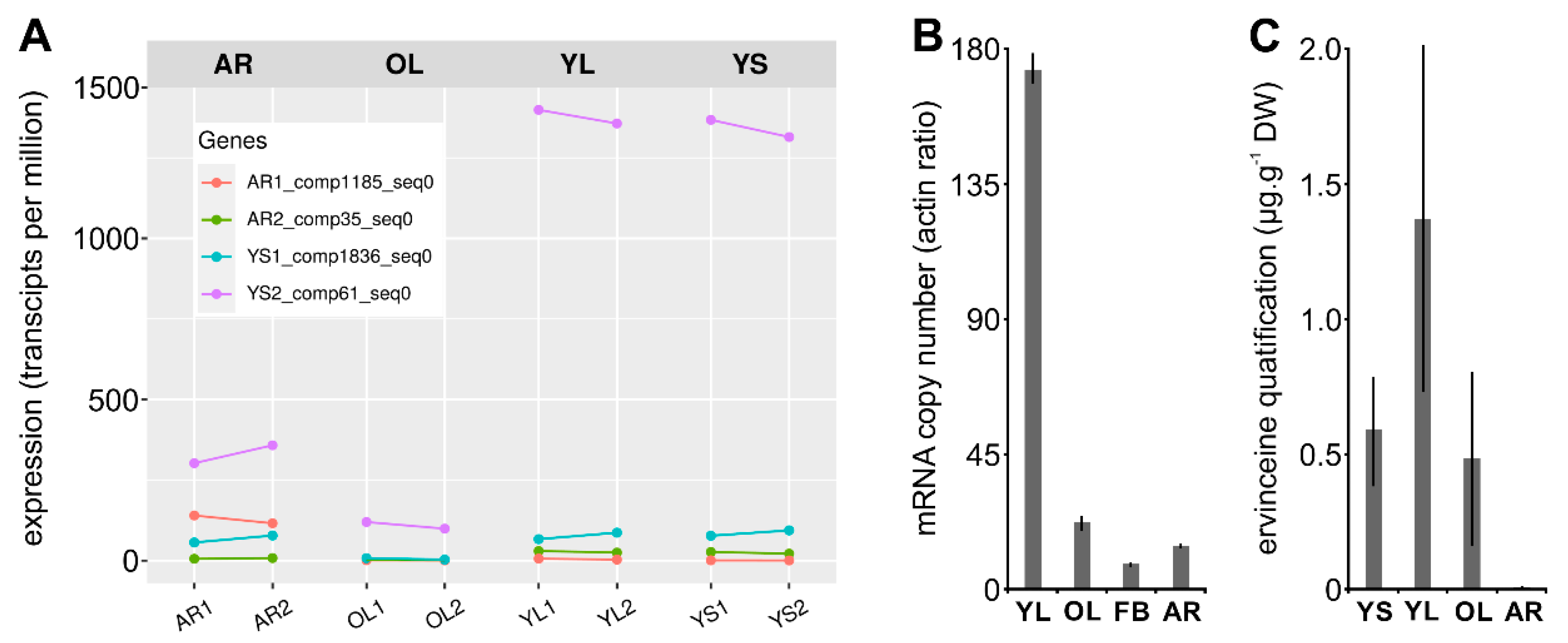
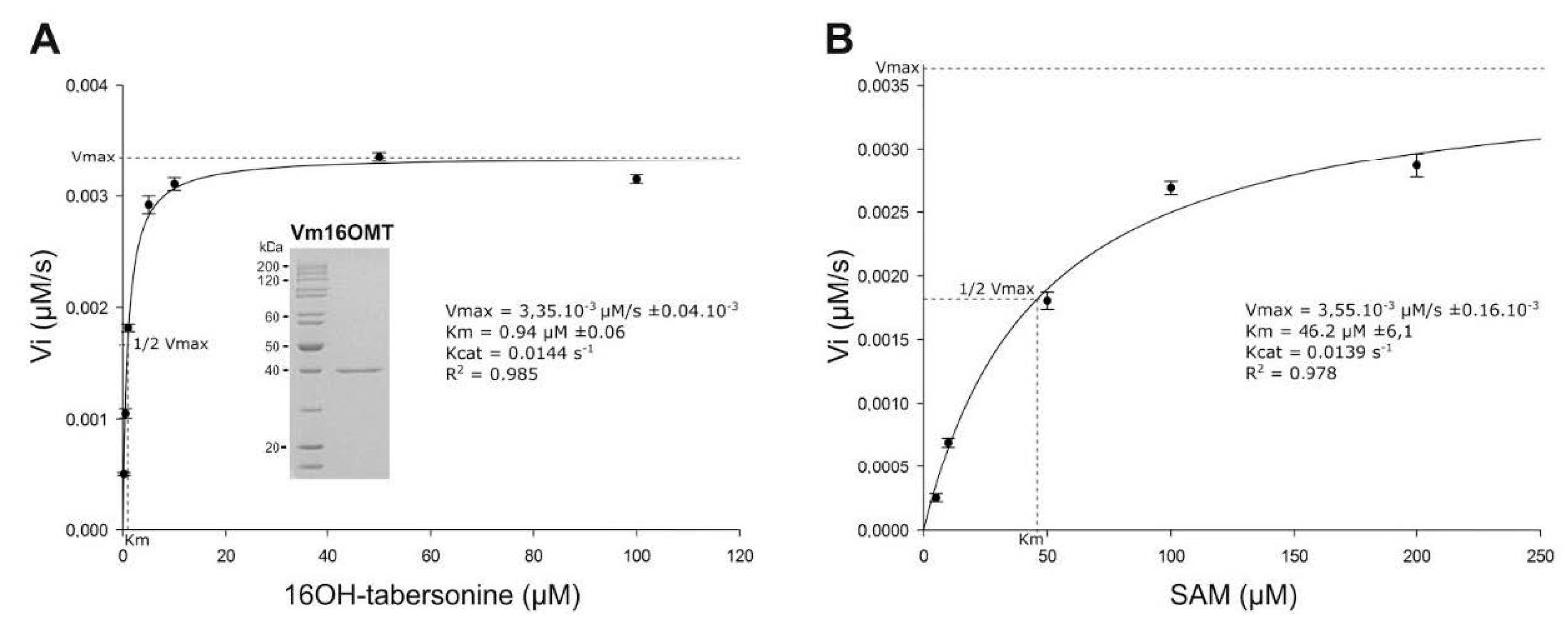
3.5. Estimation of Vm16OMT Substrate Specificity
3.6. Subcellular Localization of Vm16OMT
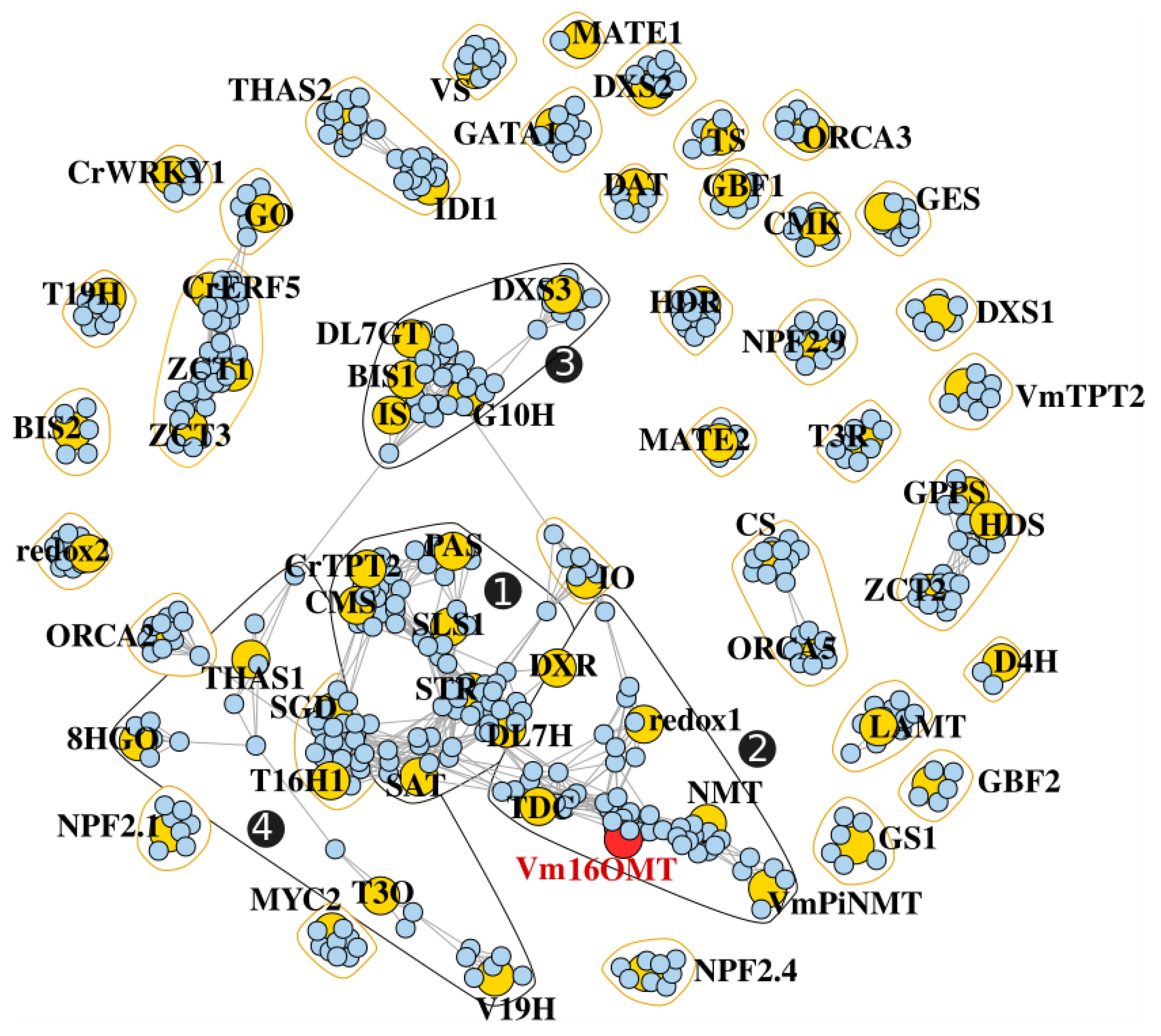
3.7. Gene Co-Expression Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luijendijk, T.J.C.; van der Meijden, E.; Verpoorte, R. Involvement of Strictosidine as a Defensive Chemical in Catharanthus roseus. J. Chem. Ecol. 1996, 22, 1355. [Google Scholar] [CrossRef]
- Roepke, J.; Salim, V.; Wu, M.; Thamm, A.M.K.; Murata, J.; Ploss, K.; Boland, W.; De Luca, V. Vinca Drug Components Accumulate Exclusively in Leaf Exudates of Madagascar Periwinkle. Proc. Natl. Acad. Sci. USA 2010, 107, 15287–15292. [Google Scholar] [CrossRef] [Green Version]
- Dugé De Bernonville, T.; Carqueijeiro, I.; Lanoue, A.; Lafontaine, F.; Sánchez Bel, P.; Liesecke, F.; Musset, K.; Oudin, A.; Glévarec, G.; Pichon, O.; et al. Folivory Elicits a Strong Defense Reaction in Catharanthus roseus: Metabolomic and Transcriptomic Analyses Reveal Distinct Local and Systemic Responses. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schläger, S.; Dräger, B. Exploiting Plant Alkaloids. Curr. Opin. Biotechnol. 2016, 37, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Carqueijeiro, I.; Langley, C.; Grzech, D.; Koudounas, K.; Papon, N.; O’Connor, S.E.; Courdavault, V. Beyond the Semi-Synthetic Artemisinin: Metabolic Engineering of Plant-Derived Anti-Cancer Drugs. Curr. Opin. Biotechnol. 2020, 65, 17–24. [Google Scholar] [CrossRef]
- Courdavault, V.; O’Connor, S.E.; Oudin, A.; Besseau, S.; Papon, N. Towards the Microbial Production of Plant-Derived Anticancer Drugs. Trends Cancer 2020, 6, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Farahanikia, B.; Akbarzadeh, T.; Jahangirzadeh, A.; Yassa, N.; Shams Ardekani, M.R.; Mirnezami, T.; Hadjiakhoondi, A.; Khanavi, M. Phytochemical Investigation of Vinca minor Cultivated in Iran. Iran. J. Pharm. Res. IJPR 2011, 10, 777–785. [Google Scholar]
- Demessie, Z.; Woolfson, K.N.; Yu, F.; Qu, Y.; De Luca, V. The ATP Binding Cassette Transporter, VmTPT2/VmABCG1, Is Involved in Export of the Monoterpenoid Indole Alkaloid, Vincamine in Vinca minor Leaves. Phytochemistry 2017, 140, 118–124. [Google Scholar] [CrossRef]
- Grossmann, E.; Šefcǒvič, P.; Szász, K. Picrinine in Vinca minor. Phytochemistry 1973, 12, 2058. [Google Scholar] [CrossRef]
- Verma, P.; Singh, N.; Khan, S.A.; Mathur, A.K.; Sharma, A.; Jamal, F. TIAs Pathway Genes and Associated MiRNA Identification in Vinca minor: Supporting Aspidosperma and Eburnamine Alkaloids Linkage via Transcriptomic Analysis. Physiol. Mol. Biol. Plants 2020, 26, 1695–1711. [Google Scholar] [CrossRef]
- Levac, D.; Cázares, P.; Yu, F.; De Luca, V. A Picrinine N-Methyltransferase Belongs to a New Family of γ-Tocopherol-Like Methyltransferases Found in Medicinal Plants That Make Biologically Active Monoterpenoid Indole Alkaloids. Plant Physiol. 2016, 170, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouzeid, S.; Beutling, U.; Surup, F.; Abdel Bar, F.M.; Amer, M.M.; Badria, F.A.; Yahyazadeh, M.; Brönstrup, M.; Selmar, D. Treatment of Vinca minor Leaves with Methyl Jasmonate Extensively Alters the Pattern and Composition of Indole Alkaloids. J. Nat. Prod. 2017, 80, 2905–2909. [Google Scholar] [CrossRef] [PubMed]
- Murata, J.; Luca, V.D. Localization of Tabersonine 16-Hydroxylase and 16-OH Tabersonine-16-O-Methyltransferase to Leaf Epidermal Cells Defines Them as a Major Site of Precursor Biosynthesis in the Vindoline Pathway in Catharanthus roseus. Plant J. 2005, 44, 581–594. [Google Scholar] [CrossRef]
- Murata, J.; Roepke, J.; Gordon, H.; De Luca, V. The Leaf Epidermome of Catharanthus roseus Reveals Its Biochemical Specialization. Plant Cell 2008, 20, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levac, D.; Murata, J.; Kim, W.S.; De Luca, V. Application of Carborundum Abrasion for Investigating the Leaf Epidermis: Molecular Cloning of Catharanthus roseus 16-Hydroxytabersonine-16-O-Methyltransferase. Plant J. 2008, 53, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Proksa, B.; Grossmann, E. High Performance Liquid Chromatographic Determination of Alkaloids from Vinca minor L. Phytochem. Anal. 1991, 2, 74–76. [Google Scholar] [CrossRef]
- O’Connor, S.E.; Maresh, J.J. Chemistry and Biology of Monoterpene Indole Alkaloid Biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar] [CrossRef]
- Salim, V.; De Luca, V. Chapter One-Towards Complete Elucidation of Monoterpene Indole Alkaloid Biosynthesis Pathway: Catharanthus roseus as a Pioneer System. In Advances in Botanical Research; Giglioli-Guivarc’h, N., Ed.; New Light on Alkaloid Biosynthesis and Future Prospects; Academic Press: Cambridge, MA, USA, 2013; Volume 68, pp. 1–37. [Google Scholar] [CrossRef]
- Dugé de Bernonville, T.; Clastre, M.; Besseau, S.; Oudin, A.; Burlat, V.; Glévarec, G.; Lanoue, A.; Papon, N.; Giglioli-Guivarc’h, N.; St-Pierre, B.; et al. Phytochemical Genomics of the Madagascar Periwinkle: Unravelling the Last Twists of the Alkaloid Engine. Phytochemistry 2015, 113, 9–23. [Google Scholar] [CrossRef]
- Thamm, A.M.K.; Qu, Y.; De Luca, V. Discovery and Metabolic Engineering of Iridoid/Secoiridoid and Monoterpenoid Indole Alkaloid Biosynthesis. Phytochem. Rev. 2016, 15, 339–361. [Google Scholar] [CrossRef]
- Caputi, L.; Franke, J.; Farrow, S.C.; Chung, K.; Payne, R.M.E.; Nguyen, T.-D.; Dang, T.-T.T.; Soares Teto Carqueijeiro, I.; Koudounas, K.; Dugé de Bernonville, T.; et al. Missing Enzymes in the Biosynthesis of the Anticancer Drug Vinblastine in Madagascar Periwinkle. Science 2018, 360, 1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laflamme, P.; St-Pierre, B.; De Luca, V. Molecular and Biochemical Analysis of a Madagascar Periwinkle Root-Specific Minovincinine-19-Hydroxy-O-Acetyltransferase. Plant Physiol. 2001, 125, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carqueijeiro, I.; Brown, S.; Chung, K.; Dang, T.-T.; Walia, M.; Besseau, S.; Dugé de Bernonville, T.; Oudin, A.; Lanoue, A.; Billet, K.; et al. Two Tabersonine 6,7-Epoxidases Initiate Lochnericine-Derived Alkaloid Biosynthesis in Catharanthus roseus. Plant Physiol. 2018, 177, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carqueijeiro, I.; Dugé de Bernonville, T.; Lanoue, A.; Dang, T.-T.; Teijaro, C.N.; Paetz, C.; Billet, K.; Mosquera, A.; Oudin, A.; Besseau, S.; et al. A BAHD Acyltransferase Catalyzing 19-O-Acetylation of Tabersonine Derivatives in Roots of Catharanthus roseus Enables Combinatorial Synthesis of Monoterpene Indole Alkaloids. Plant J. 2018, 94, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.; Qu, Y.; Simionescu, R.; De Luca, V. The Assembly of (+)-Vincadifformine- and (−)-Tabersonine-Derived Monoterpenoid Indole Alkaloids in Catharanthus roseus Involves Separate Branch Pathways. Plant J. 2019, 99, 626–636. [Google Scholar] [CrossRef]
- Kutchan, T.M.; Hampp, N.; Lottspeich, F.; Beyreuther, K.; Zenk, M.H. The CDNA Clone for Strictosidine Synthase from Rauvolfia serpentina DNA Sequence Determination and Expression in Escherichia coli. FEBS Lett. 1988, 237, 40–44. [Google Scholar] [CrossRef] [Green Version]
- McKnight, T.D.; Roessner, C.A.; Devagupta, R.; Scott, A.I.; Nessler, C.L. Nucleotide Sequence of a CDNA Encoding the Vacuolar Protein Strictosidine Synthase from Catharanthus roseus. Nucleic Acids Res. 1990, 18, 4939. [Google Scholar] [CrossRef] [Green Version]
- Kellner, F.; Geu-Flores, F.; Sherden, N.H.; Brown, S.; Foureau, E.; Courdavault, V.; O’Connor, S.E. Discovery of a P450-Catalyzed Step in Vindoline Biosynthesis: A Link between the Aspidosperma and Eburnamine Alkaloids. Chem. Commun. 2015, 51, 7626–7628. [Google Scholar] [CrossRef]
- De Luca, V.; Marineau, C.; Brisson, N. Molecular Cloning and Analysis of CDNA Encoding a Plant Tryptophan Decarboxylase: Comparison with Animal Dopa Decarboxylases. Proc. Natl. Acad. Sci. USA 1989, 86, 2582. [Google Scholar] [CrossRef] [Green Version]
- Goddijn, O.J.M.; Lohman, F.P.; de Kam, R.J.; hilperoort, R.A.; Hoge, J.H.C. Nucleotide Sequence of the Tryptophan Decarboxylase Gene of Catharanthus roseus and Expression of Tdc-GusA Gene Fusions in Nicotiana tabacum. Mol. Gen. Genet. MGG 1994, 242, 217–225. [Google Scholar] [CrossRef]
- Veau, B.; Courtois, M.; Oudin, A.; Chénieux, J.-C.; Rideau, M.; Clastre, M. Cloning and Expression of CDNAs Encoding Two Enzymes of the MEP Pathway in Catharanthus roseus. Biochim. Biophys. Acta 2000, 1517, 159–163. [Google Scholar] [CrossRef]
- Oudin, A.; Mahroug, S.; Courdavault, V.; Hervouet, N.; Zelwer, C.; Rodríguez-Concepción, M.; St-Pierre, B.; Burlat, V. Spatial Distribution and Hormonal Regulation of Gene Products from Methyl Erythritol Phosphate and Monoterpene-Secoiridoid Pathways in Catharanthus roseus. Plant Mol. Biol. 2007, 65, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Guirimand, G.; Guihur, A.; Phillips, M.A.; Oudin, A.; Glévarec, G.; Melin, C.; Papon, N.; Clastre, M.; St-Pierre, B.; Rodríguez-Concepción, M.; et al. A Single Gene Encodes Isopentenyl Diphosphate Isomerase Isoforms Targeted to Plastids, Mitochondria and Peroxisomes in Catharanthus roseus. Plant Mol. Biol. 2012, 79, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Smita, S.S.; Singh, A.K.; Shanker, K.; Nagegowda, D.A. Heteromeric and Homomeric Geranyl Diphosphate Synthases from Catharanthus roseus and Their Role in Monoterpene Indole Alkaloid Biosynthesis. Mol. Plant 2013, 6, 1531–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirimand, G.; Guihur, A.; Perello, C.; Phillips, M.; Mahroug, S.; Oudin, A.; Dugé de Bernonville, T.; Besseau, S.; Lanoue, A.; Giglioli-Guivarc’h, N.; et al. Cellular and Subcellular Compartmentation of the 2C-Methyl-D-Erythritol 4-Phosphate Pathway in the Madagascar Periwinkle. Plants 2020, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simkin, A.J.; Miettinen, K.; Claudel, P.; Burlat, V.; Guirimand, G.; Courdavault, V.; Papon, N.; Meyer, S.; Godet, S.; St-Pierre, B.; et al. Characterization of the Plastidial Geraniol Synthase from Madagascar Periwinkle Which Initiates the Monoterpenoid Branch of the Alkaloid Pathway in Internal Phloem Associated Parenchyma. Phytochemistry 2013, 85, 36–43. [Google Scholar] [CrossRef]
- Collu, G.; Unver, N.; Peltenburg-Looman, A.M.G.; van der Heijden, R.; Verpoorte, R.; Memelink, J. Geraniol 10-Hydroxylase1, a Cytochrome P450 Enzyme Involved in Terpenoid Indole Alkaloid Biosynthesis. FEBS Lett. 2001, 508, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; et al. The Seco-Iridoid Pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [Google Scholar] [CrossRef] [Green Version]
- Geu-Flores, F.; Sherden, N.H.; Courdavault, V.; Burlat, V.; Glenn, W.S.; Wu, C.; Nims, E.; Cui, Y.; O’Connor, S.E. An Alternative Route to Cyclic Terpenes by Reductive Cyclization in Iridoid Biosynthesis. Nature 2012, 492, 138–142. [Google Scholar] [CrossRef]
- Salim, V.; Wiens, B.; Masada-Atsumi, S.; Yu, F.; De Luca, V. 7-Deoxyloganetic Acid Synthase Catalyzes a Key 3 Step Oxidation to Form 7-Deoxyloganetic Acid in Catharanthus roseus Iridoid Biosynthesis. Phytochemistry 2014, 101, 23–31. [Google Scholar] [CrossRef]
- Asada, K.; Salim, V.; Masada-Atsumi, S.; Edmunds, E.; Nagatoshi, M.; Terasaka, K.; Mizukami, H.; De Luca, V. A 7-Deoxyloganetic Acid Glucosyltransferase Contributes a Key Step in Secologanin Biosynthesis in Madagascar Periwinkle. Plant Cell 2013, 25, 4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, V.; Yu, F.; Altarejos, J.; De Luca, V. Virus-Induced Gene Silencing Identifies Catharanthus roseus 7-Deoxyloganic Acid-7-Hydroxylase, a Step in Iridoid and Monoterpene Indole Alkaloid Biosynthesis. Plant J. 2013, 76, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Irmler, S.; Schröder, G.; St-Pierre, B.; Crouch, N.P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schröder, J. Indole Alkaloid Biosynthesis in Catharanthus roseus: New Enzyme Activities and Identification of Cytochrome P450 CYP72A1 as Secologanin Synthase. Plant J. 2000, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Luijendijk, T.J.C.; Stevens, L.H.; Verpoorte, R. Purification and Characterisation of Strictosidine β-d-Glucosidase from Catharanthus roseus Cell Suspension Cultures. Plant Physiol. Biochem. 1998, 36, 419–425. [Google Scholar] [CrossRef]
- Geerlings, A.; Ibañez, M.M.-L.; Memelink, J.; van der Heijden, R.; Verpoorte, R. Molecular Cloning and Analysis of Strictosidine β-d-Glucosidase, an Enzyme in Terpenoid Indole Alkaloid Biosynthesis in Catharanthus roseus. J. Biol. Chem. 2000, 275, 3051–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrinides, A.; Tatsis, E.C.; Foureau, E.; Caputi, L.; Kellner, F.; Courdavault, V.; O’Connor, S.E. Unlocking the Diversity of Alkaloids in Catharanthus roseus: Nuclear Localization Suggests Metabolic Channeling in Secondary Metabolism. Chem. Biol. 2015, 22, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Stavrinides, A.; Tatsis, E.C.; Caputi, L.; Foureau, E.; Stevenson, C.E.M.; Lawson, D.M.; Courdavault, V.; O’Connor, S.E. Structural Investigation of Heteroyohimbine Alkaloid Synthesis Reveals Active Site Elements That Control Stereoselectivity. Nat. Commun. 2016, 7, 12116. [Google Scholar] [CrossRef] [Green Version]
- Tatsis, E.C.; Carqueijeiro, I.; Dugé de Bernonville, T.; Franke, J.; Dang, T.-T.T.; Oudin, A.; Lanoue, A.; Lafontaine, F.; Stavrinides, A.K.; Clastre, M.; et al. A Three Enzyme System to Generate the Strychnos Alkaloid Scaffold from a Central Biosynthetic Intermediate. Nat. Commun. 2017, 8, 316. [Google Scholar] [CrossRef]
- Qu, Y.; Thamm, A.M.K.; Czerwinski, M.; Masada, S.; Kim, K.H.; Jones, G.; Liang, P.; De Luca, V. Geissoschizine Synthase Controls Flux in the Formation of Monoterpenoid Indole Alkaloids in a Catharanthus roseus Mutant. Planta 2018, 247, 625–634. [Google Scholar] [CrossRef]
- Qu, Y.; Easson, M.L.A.E.; Froese, J.; Simionescu, R.; Hudlicky, T.; De Luca, V. Completion of the Seven-Step Pathway from Tabersonine to the Anticancer Drug Precursor Vindoline and Its Assembly in Yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 6224. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, B.; De Luca, V. A Cytochrome P-450 Monooxygenase Catalyzes the First Step in the Conversion of Tabersonine to Vindoline in Catharanthus roseus. Plant Physiol. 1995, 109, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, G.; Unterbusch, E.; Kaltenbach, M.; Schmidt, J.; Strack, D.; De Luca, V.; Schröder, J. Light-Induced Cytochrome P450-Dependent Enzyme in Indole Alkaloid Biosynthesis: Tabersonine 16-Hydroxylase. FEBS Lett. 1999, 458, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Besseau, S.; Kellner, F.; Lanoue, A.; Thamm, A.M.K.; Salim, V.; Schneider, B.; Geu-Flores, F.; Höfer, R.; Guirimand, G.; Guihur, A.; et al. A Pair of Tabersonine 16-Hydroxylases Initiates the Synthesis of Vindoline in an Organ-Dependent Manner in Catharanthus roseus. Plant Physiol. 2013, 163, 1792. [Google Scholar] [CrossRef] [Green Version]
- De Luca, V.; Fernandez, J.A.; Campbell, D.; Kurz, W.G.W. Developmental Regulation of Enzymes of Indole Alkaloid Biosynthesis in Catharanthus roseus. Plant Physiol. 1988, 86, 447–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscombe, D.K.; Usera, A.R.; O’Connor, S.E. Homolog of Tocopherol C Methyltransferases Catalyzes N Methylation in Anticancer Alkaloid Biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18793. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Flota, F.; De Carolis, E.; Alarco, A.M.; De Luca, V. Molecular Cloning and Characterization of Desacetoxyvindoline-4-Hydroxylase, a 2-Oxoglutarate Dependent-Dioxygenase Involved in the Biosynthesis of Vindoline in Catharanthus roseus (L.) G. Don. Plant Mol. Biol. 1997, 34, 935–948. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, B.; Laflamme, P.; Alarco, A.-M.; De Luca, V. The Terminal O-Acetyltransferase Involved in Vindoline Biosynthesis Defines a New Class of Proteins Responsible for Coenzyme A-Dependent Acyl Transfer. Plant J. 1998, 14, 703–713. [Google Scholar] [CrossRef]
- De Luca, V.; Balsevich, J.; Tyler, R.T.; Eilert, U.; Panchuk, B.D.; Kurz, W.G.W. Biosynthesis of Indole Alkaloids: Developmental Regulation of the Biosynthetic Pathway from Tabersonine to Vindoline in Catharanthus roseus. J. Plant Physiol. 1986, 125, 147–156. [Google Scholar] [CrossRef]
- De Carolis, E.; Chan, F.; Balsevich, J.; De Luca, V. Isolation and Characterization of a 2-Oxoglutarate Dependent Dioxygenase Involved in the Second-to-Last Step in Vindoline Biosynthesis. Plant Physiol. 1990, 94, 1323–1329. [Google Scholar] [CrossRef] [Green Version]
- Giddings, L.-A.; Liscombe, D.K.; Hamilton, J.P.; Childs, K.L.; DellaPenna, D.; Buell, C.R.; O’Connor, S.E. A Stereoselective Hydroxylation Step of Alkaloid Biosynthesis by a Unique Cytochrome P450 in Catharanthus roseus. J. Biol. Chem. 2011, 286, 16751–16757. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lu, X.; Guo, X.; Guo, Q.; Li, D. Metabolomics Characterization of Two Apocynaceae Plants, Catharanthus roseus and Vinca minor, Using GC-MS and LC-MS Methods in Combination. Molecules 2017, 22, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenkert, E.; Wickberg, B. General Methods of Synthesis of Indole Alkaloids. IV. A Synthesis of Dl-Eburnamonine1, 2. J. Am. Chem. Soc. 1965, 87, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Kries, H.; O’Connor, S.E. Biocatalysts from Alkaloid Producing Plants. Curr. Opin. Chem. Biol. 2016, 31, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugé de Bernonville, T.; Papon, N.; Clastre, M.; O’Connor, S.E.; Courdavault, V. Identifying Missing Biosynthesis Enzymes of Plant Natural Products. Trends Pharmacol. Sci. 2020, 41, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Parage, C.; Foureau, E.; Kellner, F.; Burlat, V.; Mahroug, S.; Lanoue, A.; de Bernonville, T.D.; Londono, M.A.; Carqueijeiro, I.; Oudin, A.; et al. Class II Cytochrome P450 Reductase Governs the Biosynthesis of Alkaloids. Plant Physiol. 2016, 172, 1563–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.; Clastre, M.; Courdavault, V.; O’Connor, S.E. De Novo Production of the Plant-Derived Alkaloid Strictosidine in Yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 3205–3210. [Google Scholar] [CrossRef] [Green Version]
- Pyne, M.E.; Kevvai, K.; Grewal, P.S.; Narcross, L.; Choi, B.; Bourgeois, L.; Dueber, J.E.; Martin, V.J.J. A Yeast Platform for High-Level Synthesis of Tetrahydroisoquinoline Alkaloids. Nat. Commun. 2020, 11, 3337. [Google Scholar] [CrossRef]
- Zhao, S.; Andrade, R.B. Domino Michael/Mannich/N-Alkylation Route to the Tetrahydrocarbazole Framework of Aspidosperma Alkaloids: Concise Total Syntheses of (−)-Aspidospermidine, (−)-Tabersonine, and (−)-Vincadifformine. J. Am. Chem. Soc. 2013, 135, 13334–13337. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.; Cramer, C.L.; Huang, X. Bridger: A New Framework for de Novo Transcriptome Assembly Using RNA-Seq Data. Genome Biol. 2015, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq: Reference Generation and Analysis with Trinity. Nat. Protoc. 2013, 8. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; Vienna, Austria, 2013. Available online: https://www.r-project.org/ (accessed on 24 November 2020).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H.G. gplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Guirimand, G.; Burlat, V.; Oudin, A.; Lanoue, A.; St-Pierre, B.; Courdavault, V. Optimization of the Transient Transformation of Catharanthus roseus Cells by Particle Bombardment and Its Application to the Subcellular Localization of Hydroxymethylbutenyl 4-Diphosphate Synthase and Geraniol 10-Hydroxylase. Plant Cell Rep. 2009, 28, 1215–1234. [Google Scholar] [CrossRef] [PubMed]
- Waadt, R.; Kudla, J. In Planta Visualization of Protein Interactions Using Bimolecular Fluorescence Complementation (BiFC). Cold Spring Harb. Protoc. 2008, 2008, pdb.prot4995. [Google Scholar] [CrossRef] [PubMed]
- Guirimand, G.; Guihur, A.; Ginis, O.; Poutrain, P.; Héricourt, F.; Oudin, A.; Lanoue, A.; St-Pierre, B.; Burlat, V.; Courdavault, V. The Subcellular Organization of Strictosidine Biosynthesis in Catharanthus roseus Epidermis Highlights Several Trans-Tonoplast Translocations of Intermediate Metabolites. FEBS J. 2011, 278, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Foureau, E.; Carqueijeiro, I.; Dugé de Bernonville, T.; Melin, C.; Lafontaine, F.; Besseau, S.; Lanoue, A.; Papon, N.; Oudin, A.; Glévarec, G.; et al. Chapter Eight-Prequels to Synthetic Biology: From Candidate Gene Identification and Validation to Enzyme Subcellular Localization in Plant and Yeast Cells. In Methods in Enzymology; O’Connor, S.E., Ed.; Synthetic Biology and Metabolic Engineering in Plants and Microbes Part B: Metabolism in Plants; Academic Press: Cambridge, MA, USA, 2016; Volume 576, pp. 167–206. [Google Scholar] [CrossRef]
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.M.; Wilkins, O.; Campbell, M.M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined Sequence and Expression Comparisons across Plant Networks Derived from Seven Species. Plant Cell 2011, 23, 895–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesecke, F.; Daudu, D.; Dugé de Bernonville, R.; Besseau, S.; Clastre, M.; Courdavault, V.; de Craene, J.-O.; Crèche, J.; Giglioli-Guivarc’h, N.; Glévarec, G.; et al. Ranking Genome-Wide Correlation Measurements Improves Microarray and RNA-Seq Based Global and Targeted Co-Expression Networks. Sci. Rep. 2018, 8, 10885. [Google Scholar] [CrossRef] [Green Version]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex. Network Research. Available online: researchgate.net (accessed on 18 November 2020).
- Clauset, A.; Newman, M.E.J.; Moore, C. Finding Community Structure in Very Large Networks. Phys. Rev. E 2004, 70, 66111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, H.N.; Fragoso, V.; Paranhos, J.T.; Rau, M.R.; Fett-Neto, A.G. The Bioactive Monoterpene Indole Alkaloid N,β-d-Glucopyranosyl Vincosamide Is Regulated by Irradiance Quality and Development in Psychotria leiocarpa. Ind. Crops Prod. 2016, 86, 210–218. [Google Scholar] [CrossRef]
- Liu, Y.; Song, L.; Yu, W.; Hu, Y.; Ma, X.; Wu, J.; Ying, Y. Light Quality Modifies Camptothecin Production and Gene Expression of Biosynthesis in Camptotheca acuminata Decne Seedlings. Ind. Crops Prod. 2015, 66, 137–143. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, Y.; Chen, X.; Lee, E.J.; Barber, C.J.S.; Chakrabarty, R.; Desgagné-Penix, I.; Haslam, T.M.; Kim, Y.B.; Liu, E.; et al. Transcriptome Analysis Based on Next-Generation Sequencing of Non-Model Plants Producing Specialized Metabolites of Biotechnological Interest. J. Biotechnol. 2013, 166, 122–134. [Google Scholar] [CrossRef]
- Franke, J.; Kim, J.; Hamilton, J.P.; Zhao, D.; Pham, G.M.; Wiegert-Rininger, K.; Crisovan, E.; Newton, L.; Vaillancourt, B.; Tatsis, E.; et al. Gene Discovery in Gelsemium Highlights Conserved Gene Clusters in Monoterpene Indole Alkaloid Biosynthesis. ChemBioChem 2019, 20, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Hamilton, J.P.; Pham, G.M.; Crisovan, E.; Wiegert-Rininger, K.; Vaillancourt, B.; DellaPenna, D.; Robin Buell, C. De Novo Genome Assembly of Camptotheca acuminata, a Natural Source of the Anti-Cancer Compound Camptothecin. GigaScience 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courdavault, V.; Papon, N.; Clastre, M.; Giglioli-Guivarc’h, N.; St-Pierre, B.; Burlat, V. A Look inside an Alkaloid Multisite Plant: The Catharanthus Logistics. Curr. Opin. Plant Biol. 2014, 19, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Proksa, B.; Uhrín, D.; Grossmann, E.; Votický, Z.; Fuska, J. Relative Configuration and Cytotoxic Activity of Vincarubine: A Novel Bisindole Alkaloid from Vinca minor. Planta Med. 1988, 54, 214–218. [Google Scholar] [CrossRef]
- Lichman, B.R.; Godden, G.T.; Buell, C.R. Gene and Genome Duplications in the Evolution of Chemodiversity: Perspectives from Studies of Lamiaceae. Curr. Opin. Plant. Biol. 2020, 55, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Duplais, C.; Papon, N.; Courdavault, V. Tracking the Origin and Evolution of Plant Metabolites. Trends Plant Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zubieta, C.; He, X.-Z.; Dixon, R.A.; Noel, J.P. Structures of Two Natural Product Methyltransferases Reveal the Basis for Substrate Specificity in Plant O-Methyltransferases. Nat. Struct. Biol. 2001, 8, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Weis, K. Regulating Access to the Genome: Nucleocytoplasmic Transport throughout the Cell Cycle. Cell 2003, 112, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Oliver, S. Guilt-by-association goes global. Nature 2000, 403, 601–603. [Google Scholar] [CrossRef]
- Salim, V.; Jones, A.D.; DellaPenna, D. Camptotheca acuminata 10-Hydroxycamptothecin O-Methyltransferase: An Alkaloid Biosynthetic Enzyme Co-Opted from Flavonoid Metabolism. Plant J. 2018, 95, 112–125. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A Success Story. Genome Biol. 2000, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Eckermann, R.; Gaich, T. The Akuammiline Alkaloids; Origin and Synthesis. Synthesis 2013, 45, 2813–2823. [Google Scholar] [CrossRef]
- Rakhimov, D.A.; Malikov, V.M.; Yagudaev, M.R.; Yunusov, S.Y. Structures of Ervinceine, Ervamicine, and Ervincinine. Chem. Nat. Compd. 1970, 6, 221–224. [Google Scholar] [CrossRef]
- Van Moerkercke, A.; Steensma, P.; Schweizer, F.; Pollier, J.; Gariboldi, I.; Payne, R.; Vanden Bossche, R.; Miettinen, K.; Espoz, J.; Purnama, P.C.; et al. The BHLH Transcription Factor BIS1 Controls the Iridoid Branch of the Monoterpenoid Indole Alkaloid Pathway in Catharanthus roseus. Proc. Natl. Acad. Sci. USA 2015, 112, 8130–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Moerkercke, A.; Steensma, P.; Gariboldi, I.; Espoz, J.; Purnama, P.C.; Schweizer, F.; Miettinen, K.; Vanden Bossche, R.; De Clercq, R.; Memelink, J.; et al. The Basic Helix-Loop-Helix Transcription Factor BIS2 Is Essential for Monoterpenoid Indole Alkaloid Production in the Medicinal Plant Catharanthus roseus. Plant J. 2016, 88, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stander, E.A.; Sepúlveda, L.J.; Dugé de Bernonville, T.; Carqueijeiro, I.; Koudounas, K.; Lemos Cruz, P.; Besseau, S.; Lanoue, A.; Papon, N.; Giglioli-Guivarc’h, N.; et al. Identifying Genes Involved in Alkaloid Biosynthesis in Vinca minor through Transcriptomics and Gene Co-Expression Analysis. Biomolecules 2020, 10, 1595. https://doi.org/10.3390/biom10121595
Stander EA, Sepúlveda LJ, Dugé de Bernonville T, Carqueijeiro I, Koudounas K, Lemos Cruz P, Besseau S, Lanoue A, Papon N, Giglioli-Guivarc’h N, et al. Identifying Genes Involved in Alkaloid Biosynthesis in Vinca minor through Transcriptomics and Gene Co-Expression Analysis. Biomolecules. 2020; 10(12):1595. https://doi.org/10.3390/biom10121595
Chicago/Turabian StyleStander, Emily Amor, Liuda Johana Sepúlveda, Thomas Dugé de Bernonville, Inês Carqueijeiro, Konstantinos Koudounas, Pamela Lemos Cruz, Sébastien Besseau, Arnaud Lanoue, Nicolas Papon, Nathalie Giglioli-Guivarc’h, and et al. 2020. "Identifying Genes Involved in Alkaloid Biosynthesis in Vinca minor through Transcriptomics and Gene Co-Expression Analysis" Biomolecules 10, no. 12: 1595. https://doi.org/10.3390/biom10121595